





Atipik Kronik Myeloid Lösemi 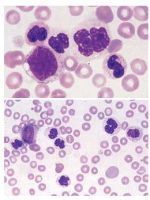 Atipik KML tanı esnasında myelodisplastik yanı sıra myeloproliferatif bulguların eşlik ettiği BCR-ABL1 negatif lösemik bir bozukluktur. Asıl olarak nötrofilik serinin tutulumu ile karakterizedir ve morfolojik olarak displastik nötrofiller ve onların prekürsörlerinin artışı ile karakterize lökositoz mevcuttur. Multilineage displazi yaygındır; bu da aKML nin stem cell kaynaklı olduğunu gösterir. Neoplastik hücreler BCR-ABL1 fuzyon genine sahip değildir. EPİDEMİYOLOJİ: Atipik KML nin gerçek insidansı bilinmemektedir. Her 100 BCR-ABL pozitif KML için yalnızca 1-2 vaka raporlanmıştır.aKML li yaşlılarda daha sıktır. Günümüzde yayınlanan birkaç seride ortalama yaş 7-8 dekatlardadır ancak daha gençlerdede bildirilmiştir. Kadın erkek oranı değişken olmakla birlikte çoğu seride 1:1 olarak bildirilmiştir. Kemik iliği ve periferik kan daima tutulur . Dalak ve karaciğer tutulumu da yaygındır. KLİNİK BULGULAR Çoğu hasta anemi ve trombositopeniye baplı semptomlara sahiptir. Diğerlerinde ise ana semptom splenomegaliye bağlı semptomlardır. MORFOLOJİ Beyaz küre sayısı her zaman 13.000 üzerindedir. Median Beyaz küre sayısı 24 bin- 96 bin olarak rapor edilmesine rağmen bazı vakalarda 300 bin üzerinde de olabilir.Blast sayısı daima %5 den azdır ve daima lökositlerin %20 sinden az olmalıdır.Nötrofil prekürsörleri genellikle %10-20 veya daha fazla olabilir. Mutlak monosit sayısı artmış olmasına rağmen monosit yüzdesi nadir olarak %10 'u geçer.Bazofili olabilir ancak baskın bir bulgu değildir. Akkiz pelger huet anomalisi ve Abnormally clumped nucleer chromatin, bizzarely segmented nucleus, abnormal cytoplasmic granularity lökositlerde diğer nükleer anormalliklerdir. Orta dereceli annemi sıktır ve eritrositlerde makroovalositler gibi diseritropoez göstergeleri görülebilir. Platelet sayısı değişkendir ancak trombositopeni sıktır.Kemik iliği biyopsisi nötrofiller ve prekürsörlerinin artmasından dolayı hiperselülerdir. Blast sayısı artmıştır ancak asla %20 yi geçmez. Nötrofilik seride disgranulopoez bulguları mevcuttur. Megakaryositler azalmış, normal veya artmış olabilir ancak dismegakaryopoez bulguları görülür.M:E oranı 10:1 den daha yüksektir. Artmış retikülin fibrozisi tanı esnasında veya hastalığın gidişatı sırasında gözlenebilir. İMMUNFENOTİP Günümüzde karakteristik immünfenotip bildirilmemiştir. İmmümohistokimyasal çalışmalarla CD14 veya CD68R biopsi spesmenlerinde pozitifliği monositleri tanımlayabilir dolayısı ile kemik iliğinde Belirgin monositoz olması acaba aKML ile karşıkarşıyamıyız sorusunu akla getirmelidir. GENETİK Atipik KML li hastaların %80 inde karyotipik anomaliler bulunur. En yaygın anormallikler +8 del20q dur. Ancak 13,14,17,19,12. kromozom anormallikleri bildirilmiştir. İzole izokromozom17 q da görülebilir.Tipik olarak BCR -ABL1 füzyon geni yoktur. Bazı vakalarda JAK2V617F tespit edilmiştir.Yaklaşık %30 vakada akkiz NRAS ve K rAS mutasyonu ile ilşkili bulunmuştur. Yine bazı olgularda t(8;9)(p22;p24) PCM1-JAK2 füzyon geni varlığı bildirilmiştir. Ancak günümüzde bu vakaların daha çok eozinofili ile seyrettiği myelodisplazinin olmadığı görülmüş ve bu vakaların daha çok kronik eozinofilik lösemiye uyduğu belirtilmiştir. PROGNOZ Bu hastalrın prognozu olduça kötüdür. Küçük sayıda hasta grupları bildirilmiş olup ortalama sağkalım 14-29 aydır. >65 yaş; Kadın Cinsiyet; WBC>50.000 ; Trombositopeni; Hb<10 gr="" dl="" k="" t="" prognoz="" fakt="" rleridir="" br="">Yaklaşık %15-40 hasta akut lösemiye döner. ölüm daha çok kemik iliği yetmezliğinden meydana gelir.
| |||||||||
|
9723 kez okundu
YorumlarHenüz yorum yapılmamış. İlk yorumu yapmak için tıklayın |